

Genetics and Metabolism
- Page Path
-
- HOME
- TOPICS
- Genetics and Metabolism
- Clinical Note
- Review Article
- Editorial
- Review Article
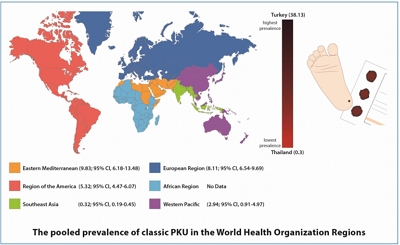
- Systematic review and meta-analysis
- Editorial
- Original Article
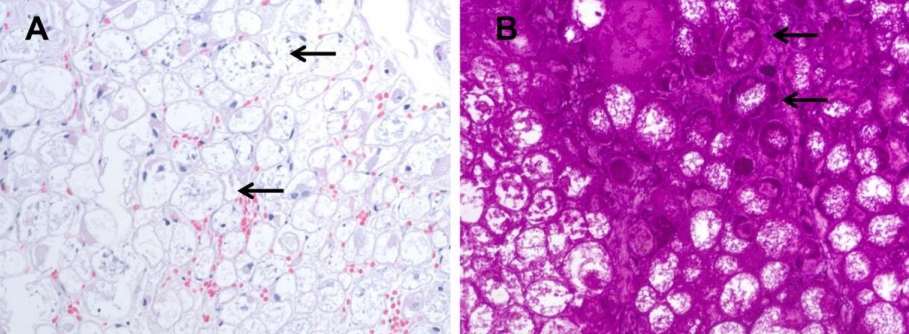
- Case Report
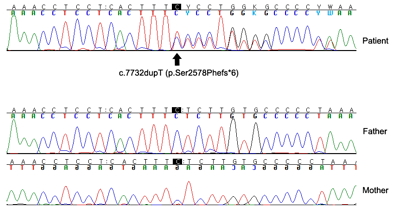
- Letter to the Editor
- Case Report
- Original Article
- Case Report
- Original Article
-

-
-

-

-
Impact Factor4.2
-
6.52022CiteScore92nd percentilePowered by




")